Download the binary distribution and expand it.
Getting started with MARICI
I expect that you will be surprised at the overwhelming speed to create prototypes of crystal structures. I hope that you agree with my curiosity and the future prospect of mathematical crystal chemistry. If you are a Windows user, graphic user interface will help you begin your MARICI journey.
Download the latest version of MARICI
How to download binary distribution of MARICI on Windows?
How to build source distribution of MARICI on Visual Studio 2026?
You can build MARICI on Visual Studio 2026 as follows: First, download the source distribution for Visual Studio 2026, and expand it on the folder of "C:/Projects/2026/C++". Note that "Marici 2026.slnx" depends on many additional include directories such as "C:/Projects/2026/C++/Library/*". Please refer to the webpage of Microsoft to understand how to customize the property of the solutions in Visual Studio 2026. Second, if you have already downloaded Visual Studio 2026, you can open the "Marici 2026.slnx" file by clicking it. Third, you will see all the "*.cpp" and ".h" files clearly, but you cannot build it if you have not yet installed Intel OneAPI Toolkit. The installation of the Toolkits is straightforward: You have only to follow the instruction of the installer, but be sure to designate Visual Studio 2026 as a native compiler. Finally, you have to link the SPGLIB library to analyze the space group of a crystal structure as follows: First, download the zip file from its website, and expand it on the folder of "C:/Libs/spglib". Second, open the cmake-gui that can be downloaded from its website, and be sure to select the source and build folder as "C:/Libs/spglib/spglib" and "C:/Libs/spglib/spglib_build", respectively. Third, please push "configure" button twice, and be sure to uncheck "SPGLIB_WITH_TEST". Fourth, please push "configure" button twice again and push "generate" button once. Fifth, please open the "Spglib.slnx", and build the solution for the Release version. Sixth, it is better to move "symspg.lib" and "symspg.dll" into "*/Marici 2026/x64/Release" folder, which is the folder for Release-built "Marici 2026". Seventh, it is important to note that you can change the folder for installing SPGLIB, but be sure to open the property of "Marici 2026.slnx" to change the additional library directory as written here. The "Marici 2026.exe" will be built without any warning and error.
How to compile MARICI on Linux?
You can compile MARICI on Linux as follows: First, download the source distribution for intel compiler on Linux, and expand it on your "Project directory" on Linux. Second, you must download the "cmake-*.*.*.tar.gz" from its website, and expand it on your "cmake directory". Third, please execute the command "./bootstrap" and "make" to install the cmake, and register the path of "*/cmake-*.*.*/bin" through the ".bashrc" file. Fourth, you must download the "spglib-*.*.*.tar.gz" from its website, and expand it on your "spglib directory". and SPGLIB library. Fifth, you can easily install it by following the instruction. Note that you must register the path of "*/spglib/lib64" in "LD_LIBRARY_PATH" through the ".bashrc" file. Sixth, be sure to copy the "libsymspg.so", which is made in the directory of "*/spglib-*.*.*/lib64", to the directory of "*/Marici", where the "Makefile" is placed. Seventh, you will make success installing "Marici.exe" after executing "Make" on the folder. It is important to note that "-std=c++20" and "-lstdc++fs" flags are necessary, and besides, you must write the frag to use MPI such as "parallel" (maybe it depends on computers). Furthermore, "-qmkl=sequential" flag is necessary if you select the source code using Intel MKL. Finally, if you have some trouble, please refer to the memo attached below for downloading the cmake and SPGLIB.
| Contents | File name |
|---|---|
| Installation of cmake and SPGLIB library on linux | install-memo.txt |
How to execute MARICI on Windows?
Click "*/Marici.exe" in the folder for binary distribution. If you receive the alert due to the missing of the ".NET 10", feel free to download it from Microsoft. It is the universal platform to execute a software, and the data is small. If you succeed in launching the graphic user interface of MARICI, please follow the instruction of MARICI. You can easily begin to predict or analyze crystal structures.
How to execute MARICI on Visual Studio 2026?
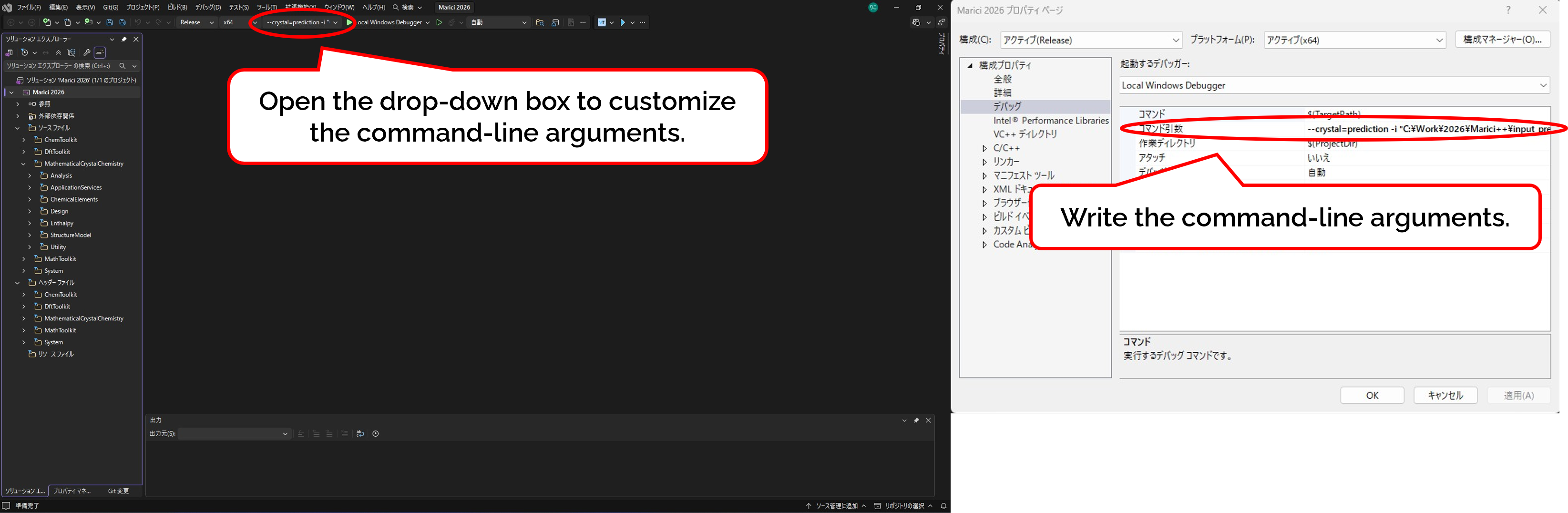
If you have already built the Release version of the "Marici 2026.exe", it is straightforward to use it. However, you have to write the appropriate command-line arguments as follows: As shown in the figure attached below, open the drop-down box to open the property box to customize command-line arguments. The command-line flags are listed below. Note that you must designate the input file as "-i */input.txt", and choose either "--crystal=prediction" or "--crystal=extraction". If you have any trouble, please refer to the batch files attached below.
| Flag | Meaning |
|---|---|
| --crystal=prediction | Selece mode to design prototypes of crystal structures. |
| --crystal=extraction | Selece mode to extract crystal structures from dataset consisting of CIF files. |
| -i */input.txt | Designate the path to an input file. |
| Execution mode | Sample batch file |
|---|---|
| Crystal structure prediction | crystal-prediction.bat |
| Crystal structure extraction | crystal-extraction.bat |
How to execute MARICI on Linux?
When executing MARICI as "srun $HOME/Marici/build/Marici.exe", you must add the command-line arguments. The flags are listed below. Note that you must designate the input file as "-i */input.txt", and choose either "--crystal=prediction" or "--crystal=extraction". If you have any trouble, please refer to the batch files attached below. While they support executing MARICI on Windows, the execution command is almost the same.
| Flag | Meaning |
|---|---|
| --crystal=prediction | Selece mode to design prototypes of crystal structures. |
| --crystal=extraction | Selece mode to extract crystal structures from dataset consisting of CIF files. |
| -i */input.txt | Designate the path to an input file. |
| Mode | Sample batch file |
|---|---|
| Crystal structure prediction | crystal-prediction.bat |
Is there any trouble to use GNU compilers?
You cannot use any mode except for "crystal structure prediction mode". You can predict crystal structures by MARICI, but you cannot extract crystal structures from the large dataset of CIF files.
Crystal structure prediction on Windows
How to copy previous parameters to design crystal structures?
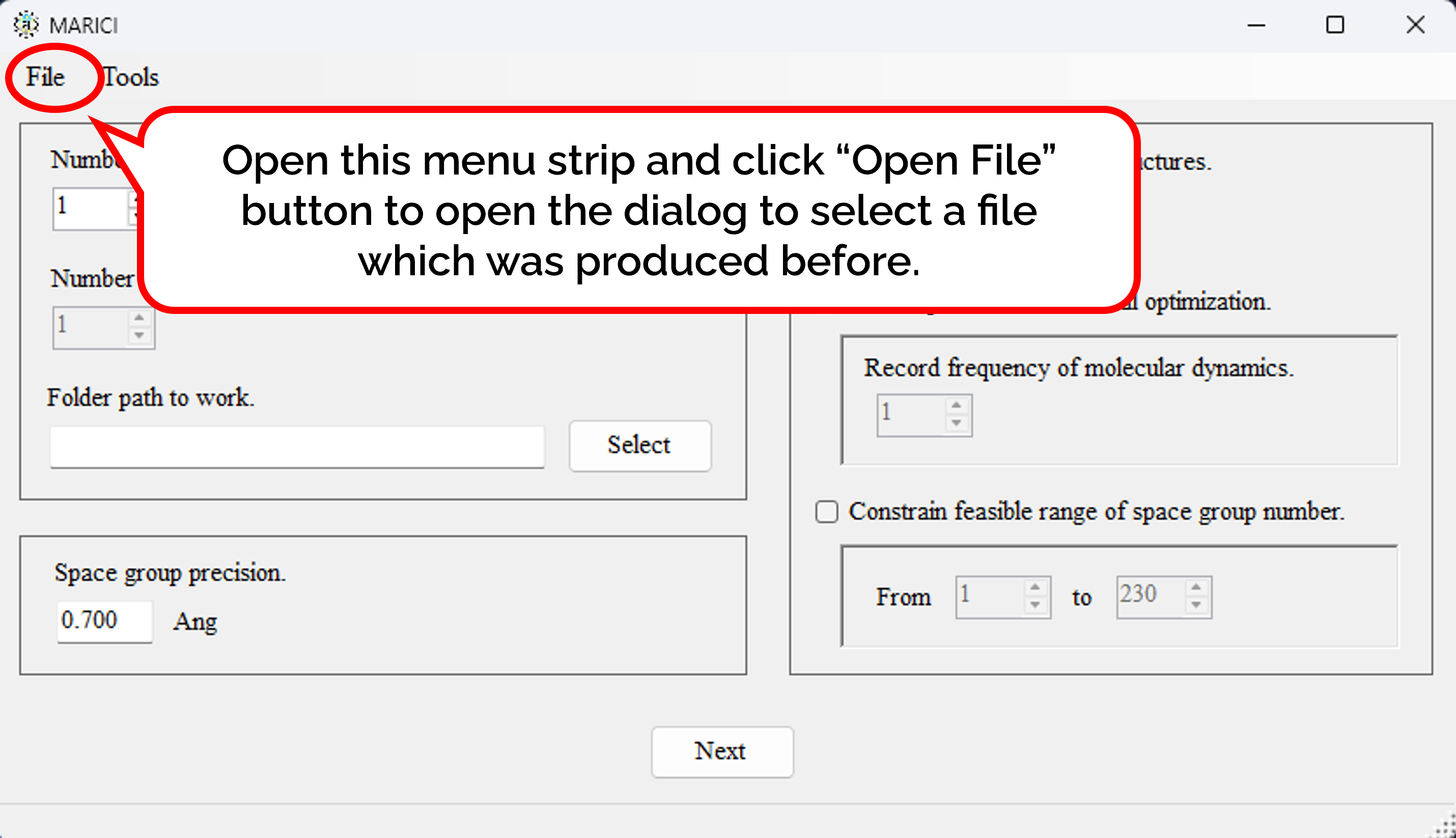
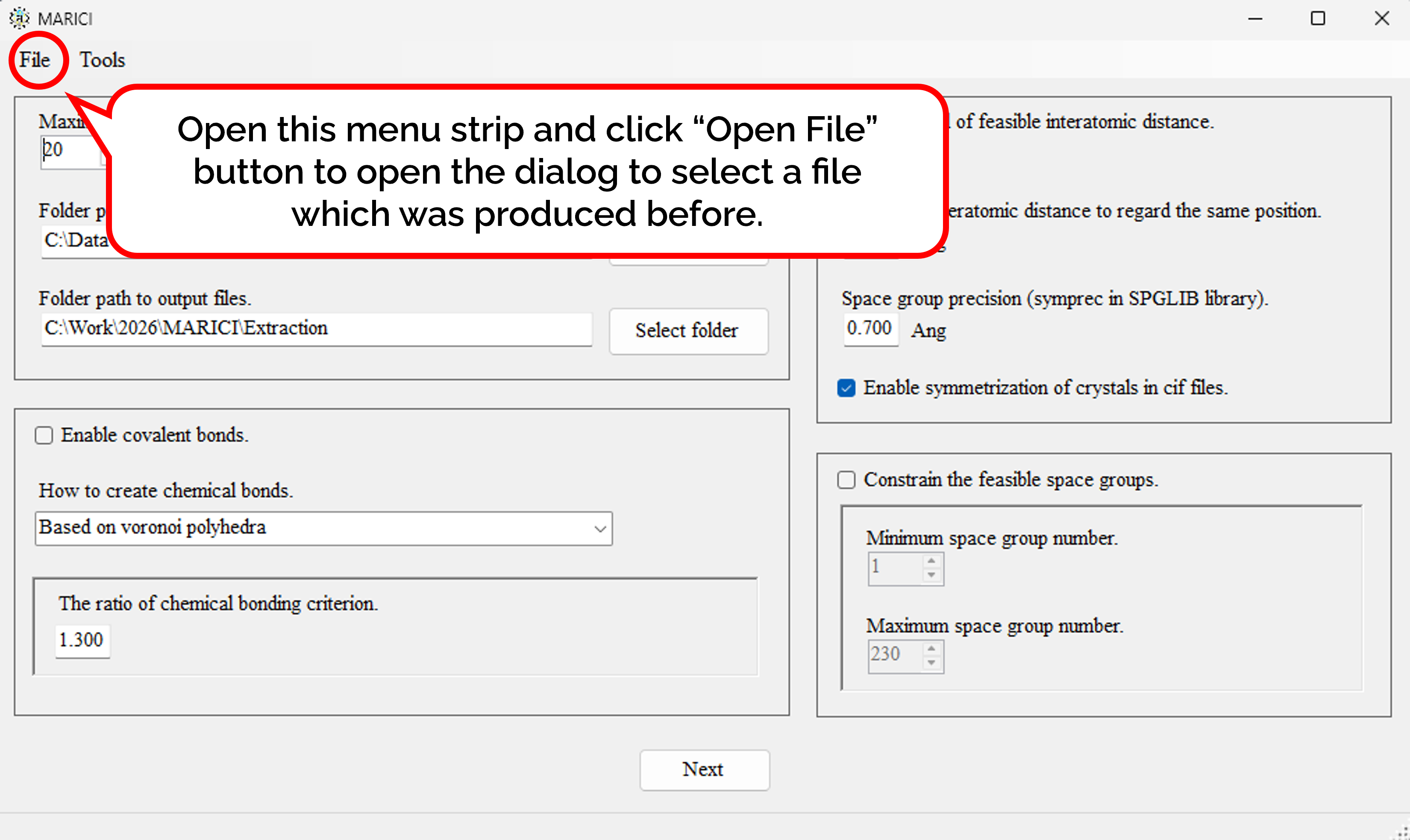
You can easily copy the previous parameters to design crystal structures as follows: First, open the menu strip named "File" and click "Open File". Second, select a file which was produced by MARICI before via a "open file dialog". Accordingly, all the parameters including computer setting are copied from the file.
| Design thema | Sample input file |
|---|---|
| Prediction of a quaternary oxide | quaternary_oxide.txt |
| Prediction of four kinds of Perovskite-type structures | perovskite_type.txt |
| Prediction of alpha-Pyrochlore and pyroxene | alpha-pyrochlore_and_pyroxene.txt |
| Predictions of Zintl phases | zintl.txt |
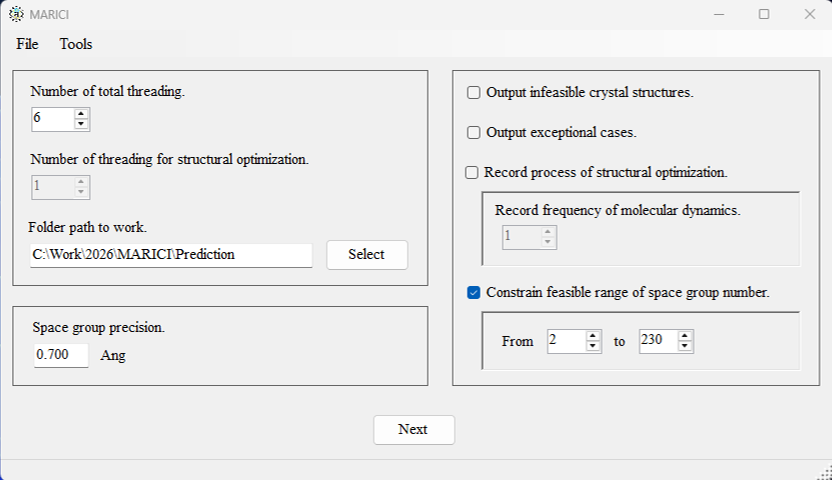
How to configure the computer setting?
You can easily configure the computer setting via the user interface attached below. Note that if you activate the records of structural optimizations, "*.md" files are generated. They can be visualized by OpenMX Viewer.
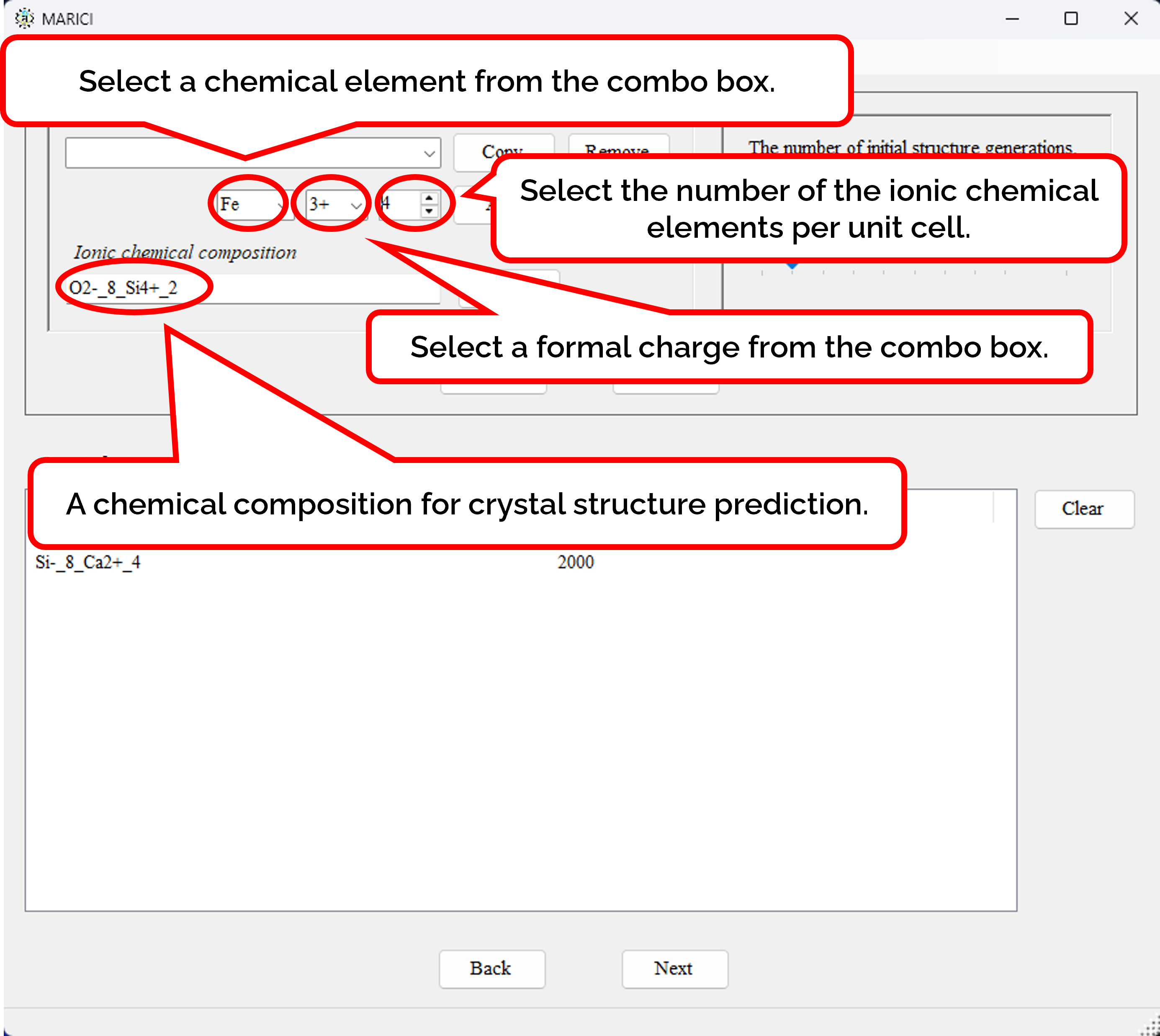
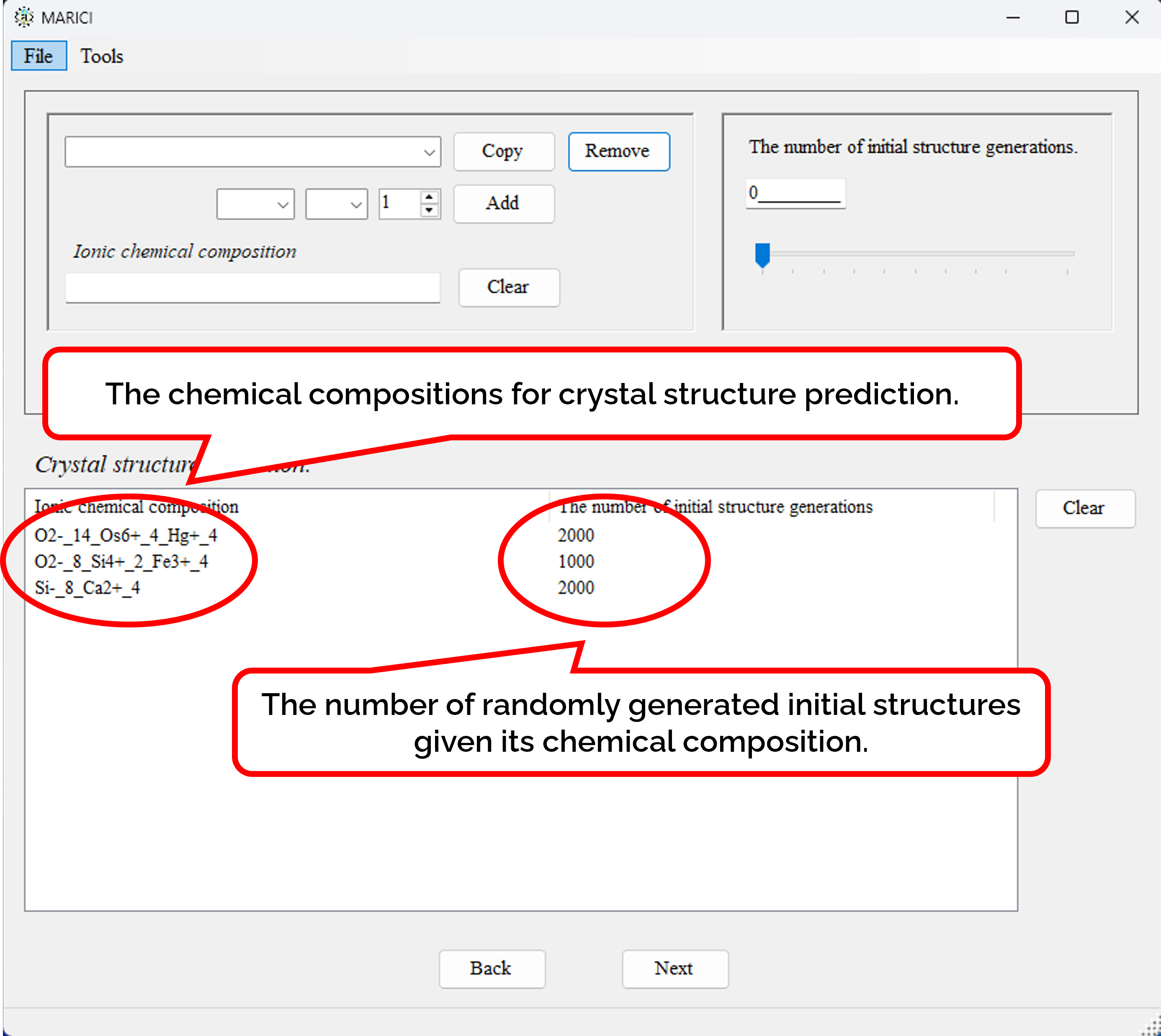
How to set the chemical compositions for crystal structure prediction?
You can easily set the chemical compositions for crystal structure prediction as shown in the figures attached below. First, customize a chemical composition by selecting chemical elements with their formal charges and the number of them per unit cell. Note that formal charges are necessary to judge whether an ionic bond are creatable or not between a pair of atoms. Second, you set the number of randomly generated initial structures given its chemical composition. If you finish making the lists including all the chemical compositions, let you go next.
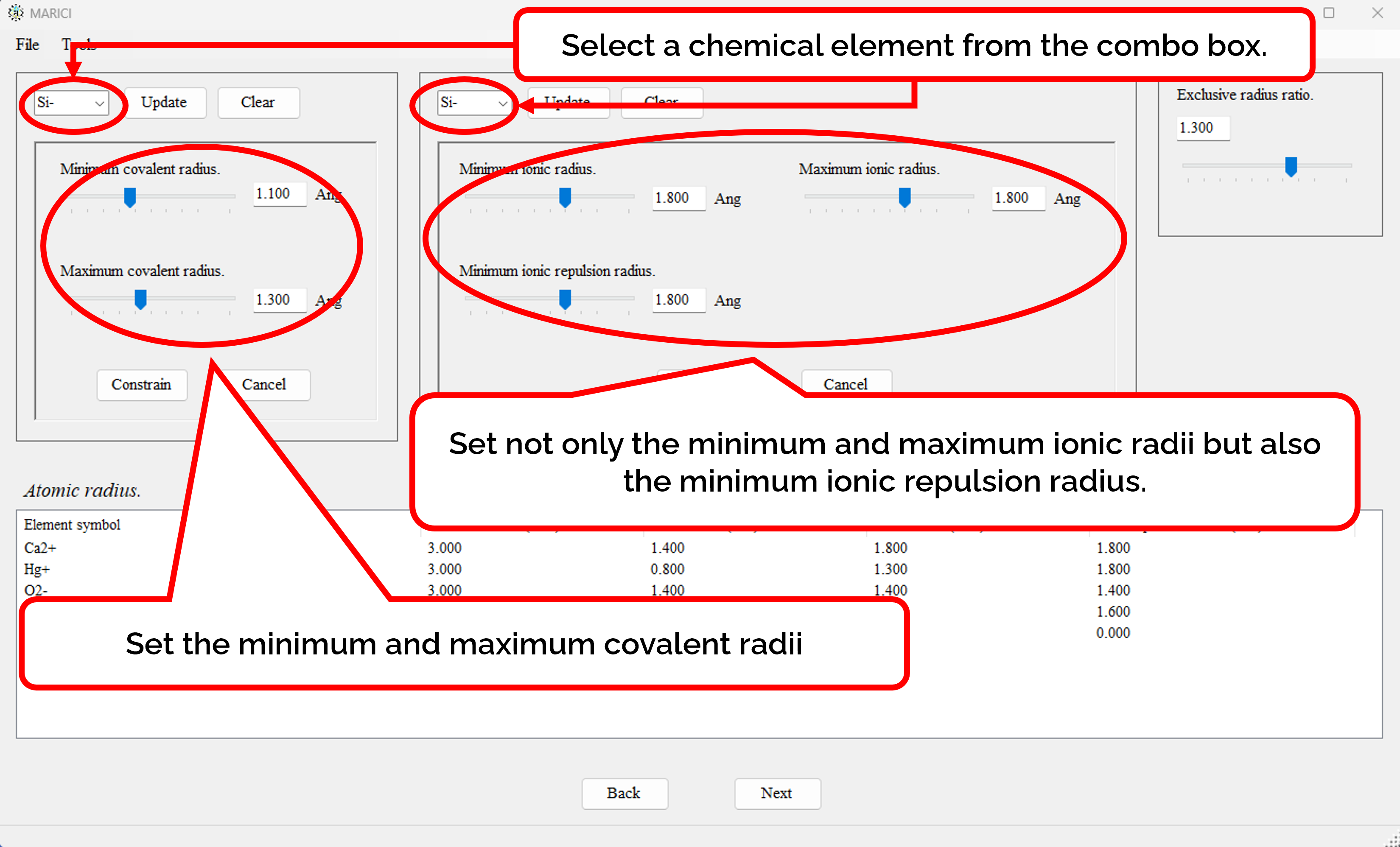
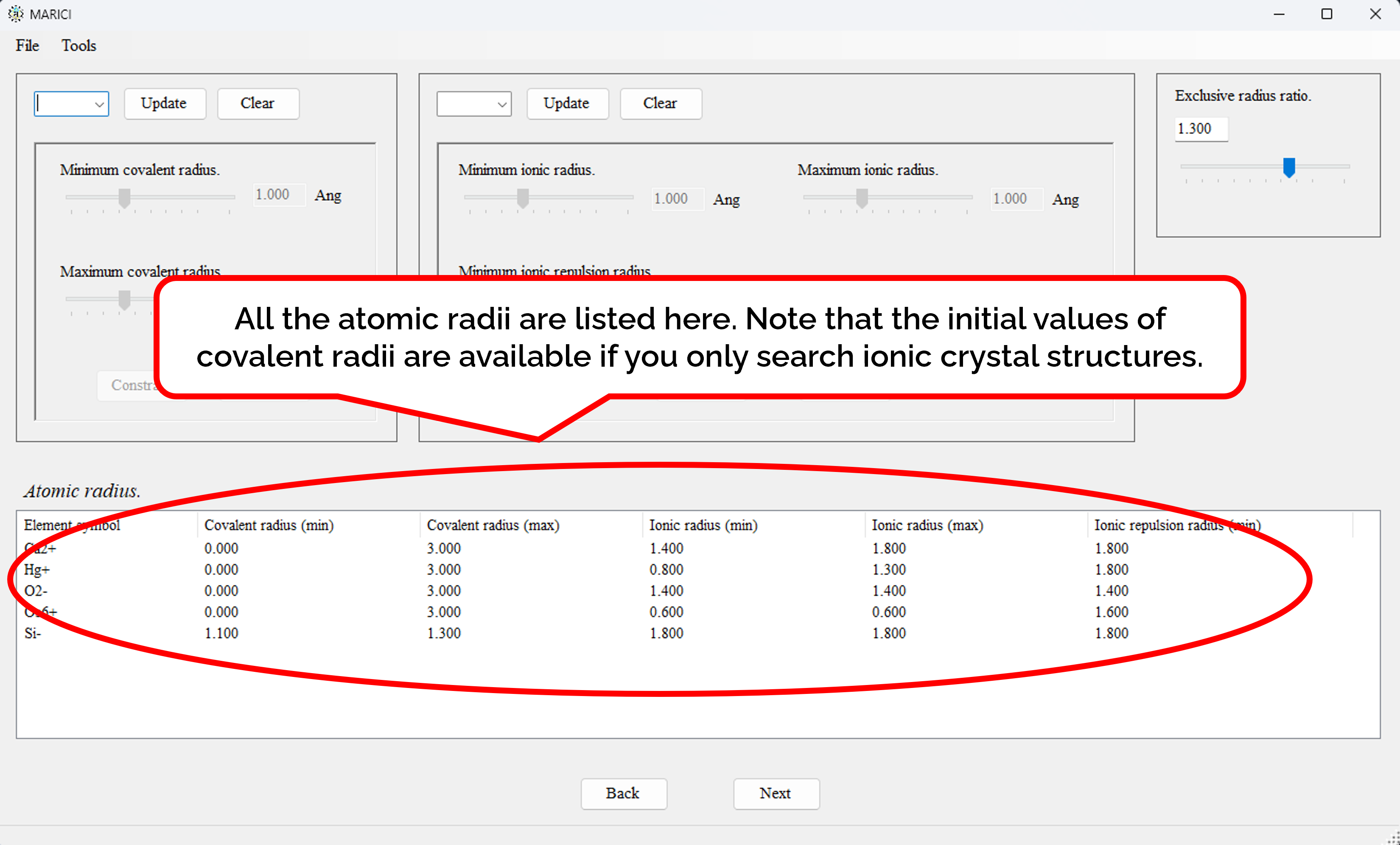
How to assign the atomic radii?
You can easily set the atomic radii of each ionic chemical element as shown in the figures attached below. First, select an ionic chemical element from the combo box. Second, assign atomic radii. Note that if you only search the crystal structures of ionic compounds, the initial values of covalent radii are available, because covalent radii will not used. If you finish the assignment, click the "constrain" button. If you finish making the lists including all the atomic radii, let you go next.
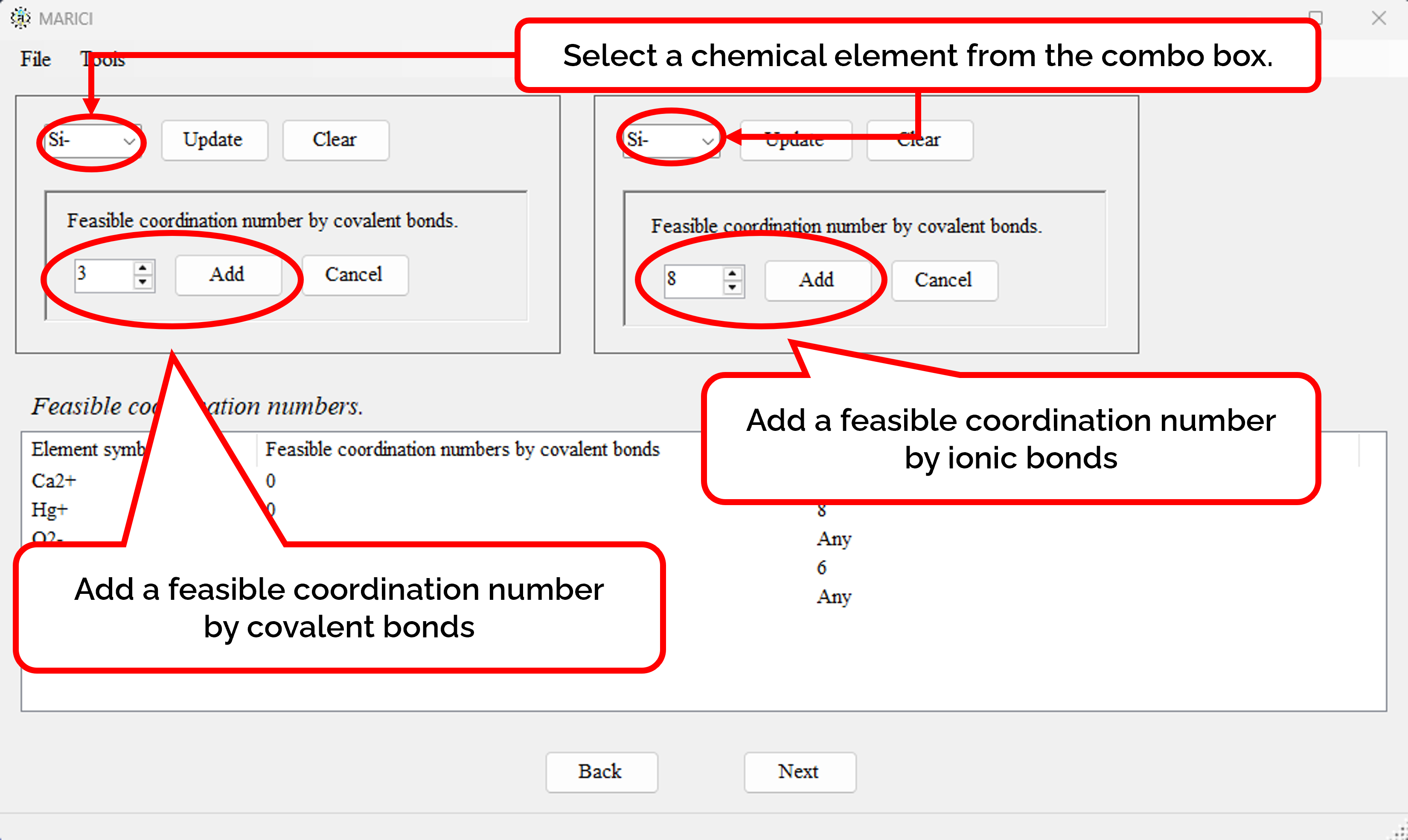
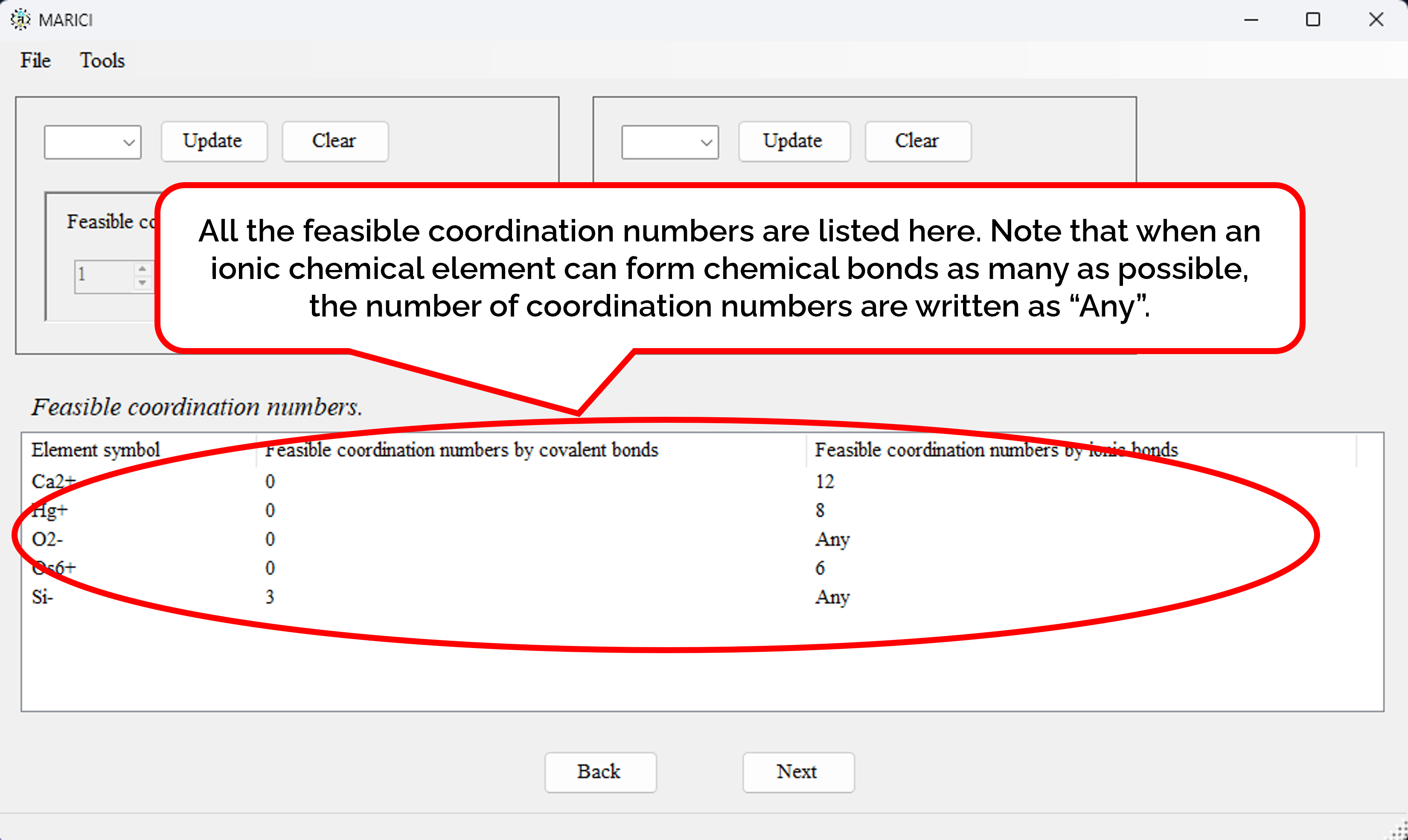
How to assign the feasible coordination numbers?
You can easily set the feasible coordination numbers of each ionic chemical element as shown in the figures attached below. First, select an ionic chemical element from the combo box. Second, add a feasible coordination number depending on the type of chemical bonds. Note that if you only search the crystal structures of ionic compounds, the the feasible coordination numbers by covalent bonds must be zero. If you finish the addition, click the "constrain" button. Note that if an ionic chemical element can form as many covalent or ionic bonds as possible, the feasible coordination number is written as "Any". If you finish making the lists including all the feasible coordination numbers, let you go next. It should be important to note that if there are no constraint on coordiantion numbers of an atom, the atom create too many chemical bonds in the algorithm of the latest MARICI. Therefore, if you search crystal structures of ionic compounds, you should constrain the coordination number of all the cations.
How to customize numerical options?
The graphic user interface supports customizing a part of numerical options for initial structure generation and structural optimization. If you are interested in the dependency of numerical options on the computational performance, you must write the input file yourself. Please see the document to learn how to write input files.
Crystal structure extraction on Windows
How to copy previous parameters to extract crystal structures?
You can easily copy the previous parameters to extract crystal structures as follows: First, open the menu strip named "File" and click "Open File". Second, select a file which was produced by MARICI before via a "open file dialog". Accordingly, all the parameters including computer setting are copied from the file.
| Extraction thema | Sample input file |
|---|---|
| Extraction of ionic compounds consisting of coordination polyhedra whose coordination numbers are 4,6, or 12. | 4-6-12_ionic_compounds.txt |
| Extraction of oxides with frustrations caused by linking of octahedra via vertex sharings | octahedral_frustration.txt |
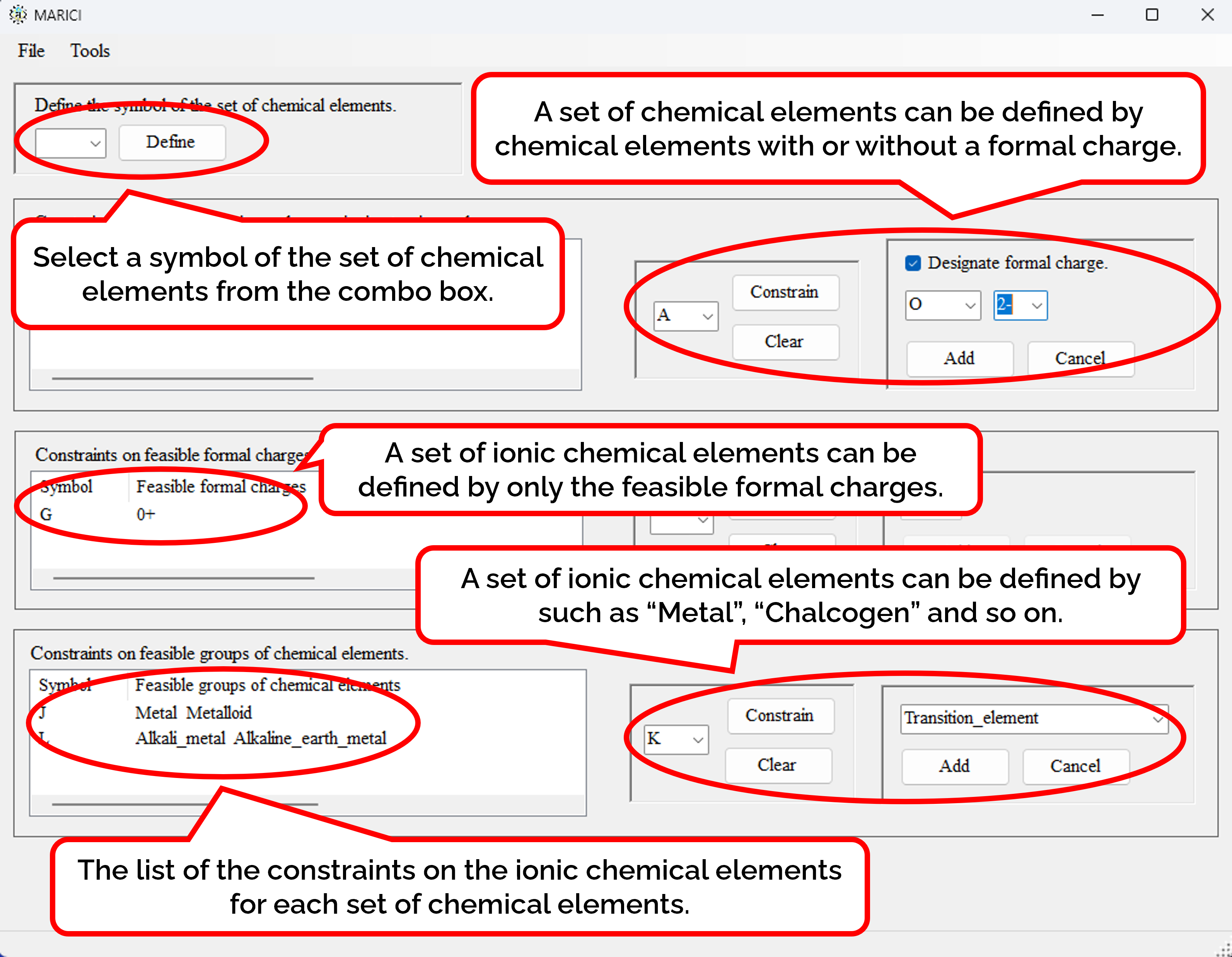
How to define the set of ionic chemical elements?
First, you have to designate the symbol of the set of ionic chemical elements from the combo box. Seconds, you can constrain the feasible ionic chemical elements of each set by not only atomic symbol but also the formal charges, and so on. When you go to the next form, you can easily constrain the infeasible ionic chemical elements in the same way.
How to constrain the feasible compositions of coordination polyhedra?
First, you can easily constrain the feasible compositions of coordination polyhedra of atoms belonging to a set of ionic chemical elements as shown in the figure attached below. Second, you can specify the type of chemical bonds, but the developer have not yet confirmed that such algorithm execute correctly. Third, you can constrain the feasible symmetries of the coordination polyhedra of atoms belonging to a set of ionic chemical elements. When you move on the next form, you can easily define the infeasibility of coordination polyhedra in the same way.

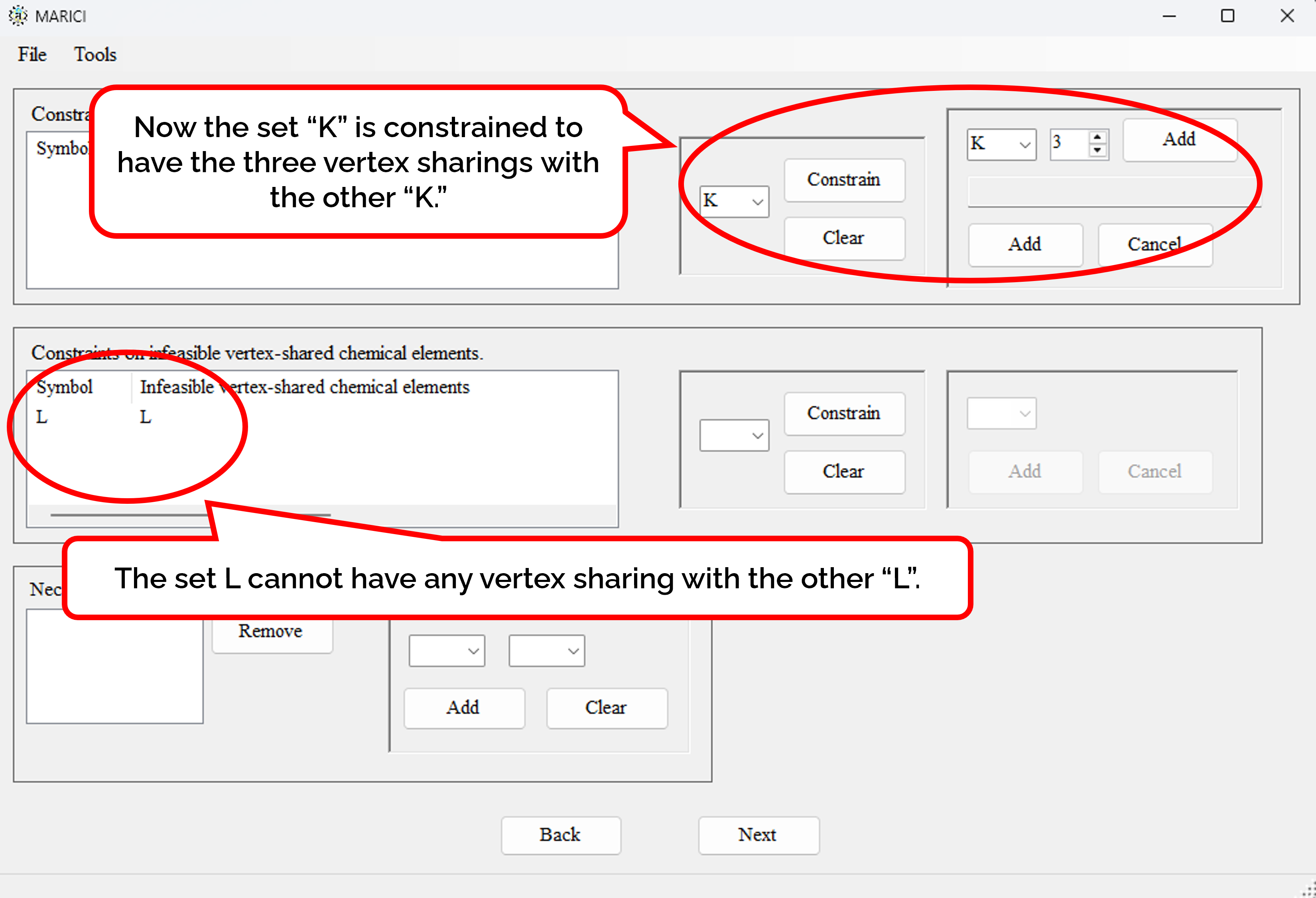
How to constrain the linkings of coordination polyhedra?
You can constrain the number of vertex sharings with a specified set of ionic chemical elements. You can also prohibit the vertex sharings with a specified set of ionic chemical elements. If you move on the next forms, you can easily make constraints on edge and face sharings in the same way.
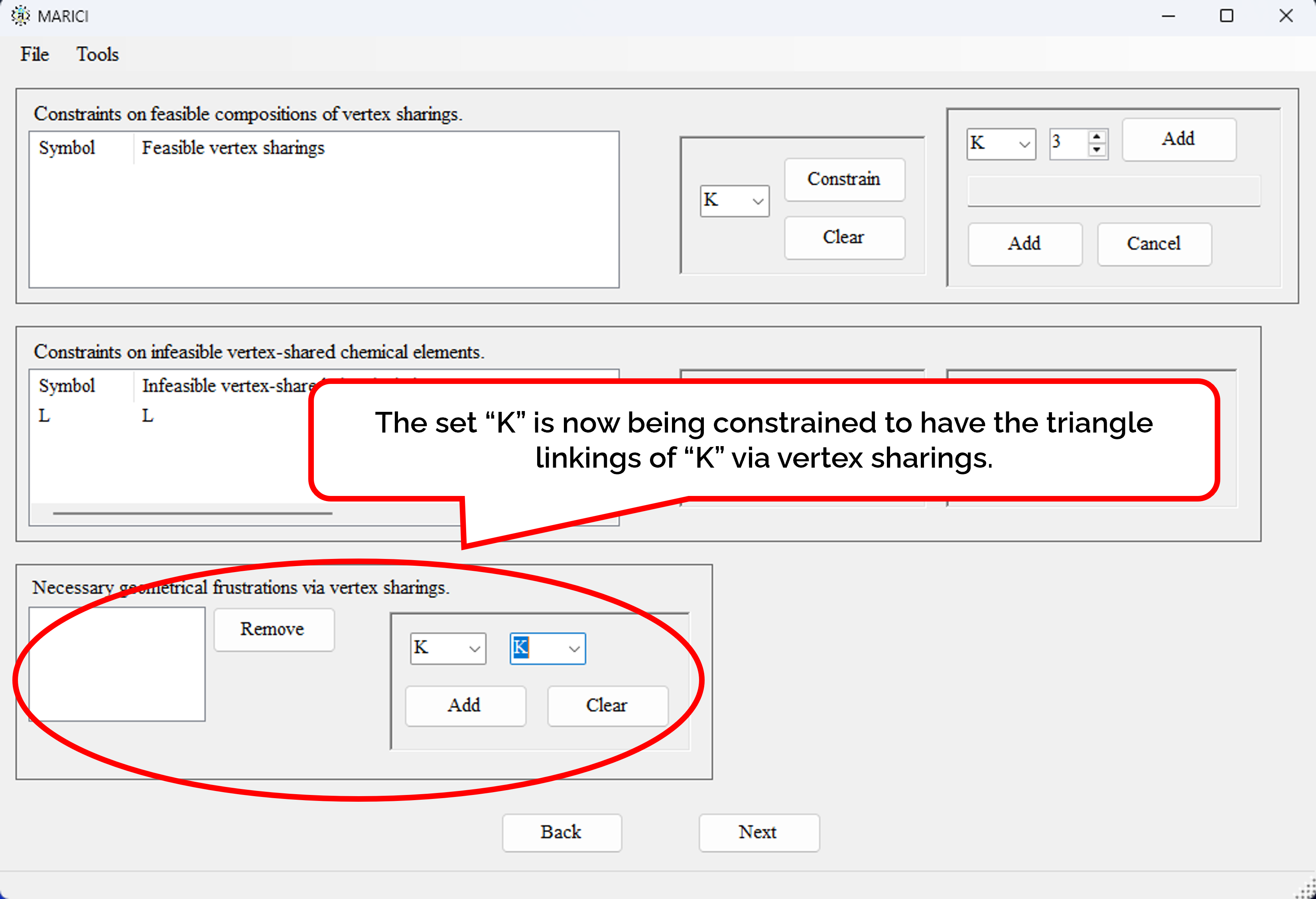
How to find the geometrical frustration?
You can find crystal structures with frustration consisting of the specified sets of ionic chemical elements via vertex sharings. Note that the frustration is defined as a triangle made by the linkings of the specified sets of ionic chemical elementss. you can also find frustrations via edge and face sharings in the same way.
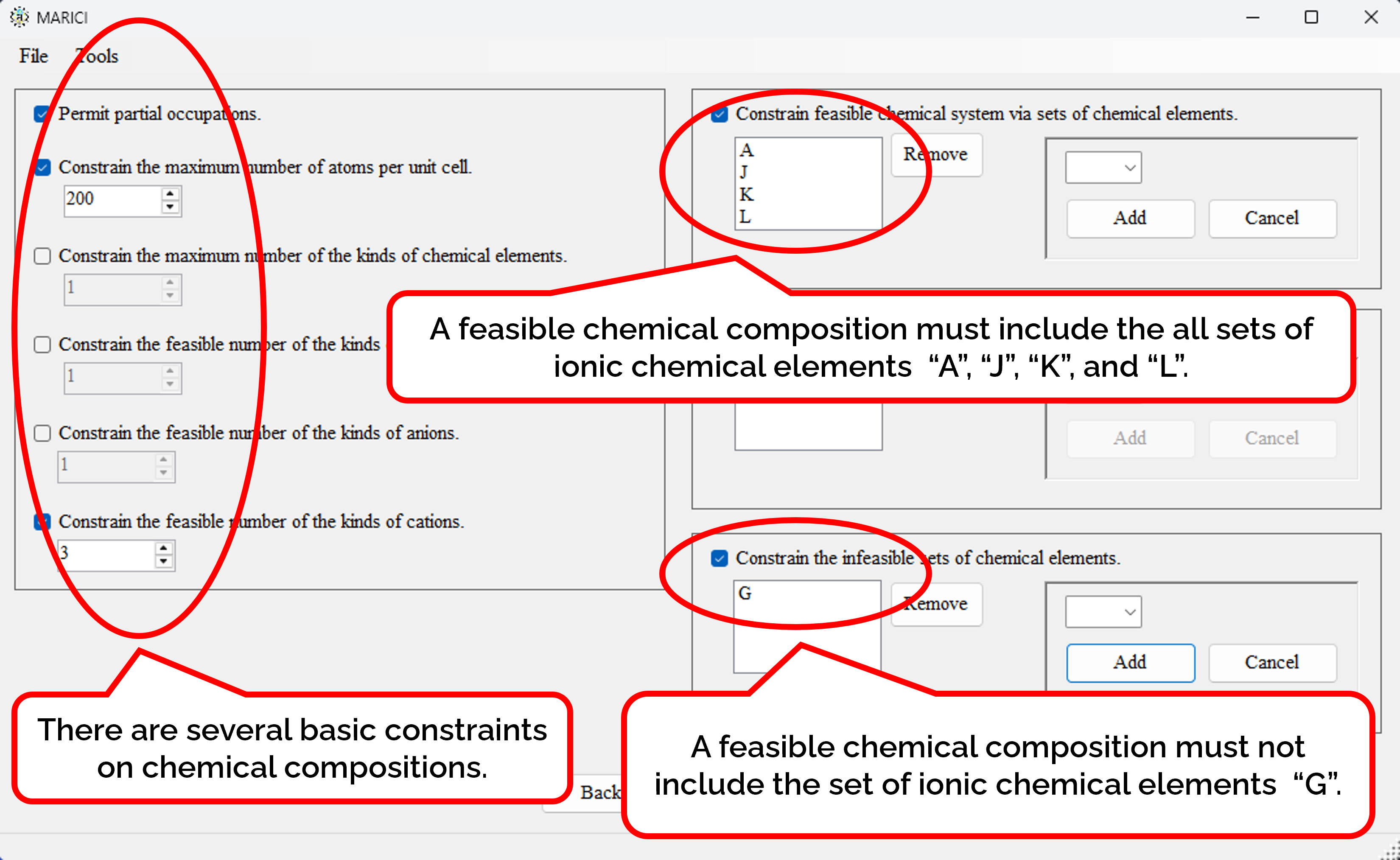
How to constrain the feasible chemical compositions?
There are several basic constraints on chemical compositions such as the maximum number of atoms per unit cell. Besides, you can constrain the feasible chemical system, which is the collection of the sets of ionic chemical elemeents. Furthermore, you can designate the necessary sets of ionic chemical elements. Finally, you can designate the infeasible sets of ionic chemical elements.
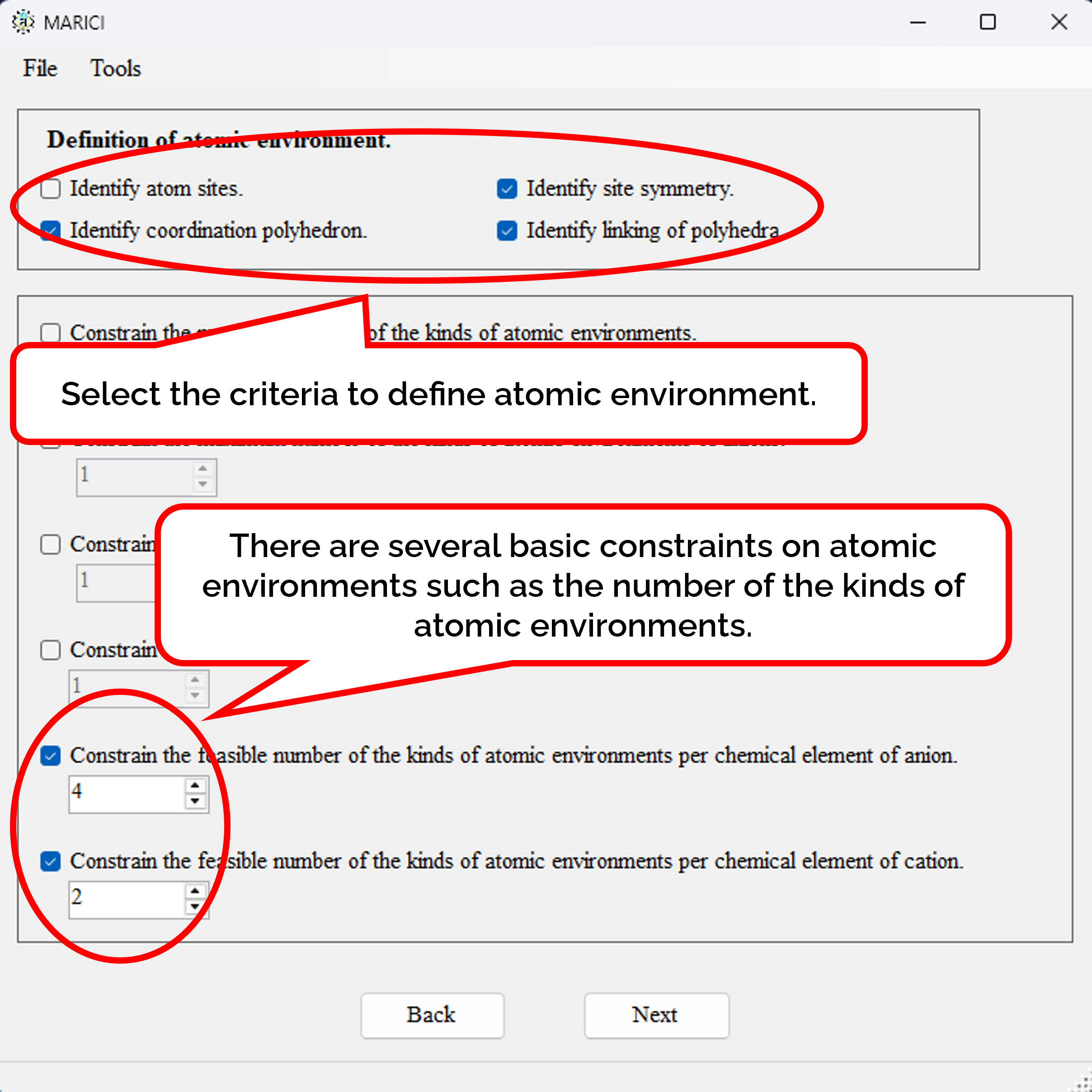
How to define and constrain the atomic environments?
You must define the atomic environments by such as the composition of coordination polyhedron. There are several constraints on the coordination polyhedra such as the maximum number of the kinds of atomic environments.
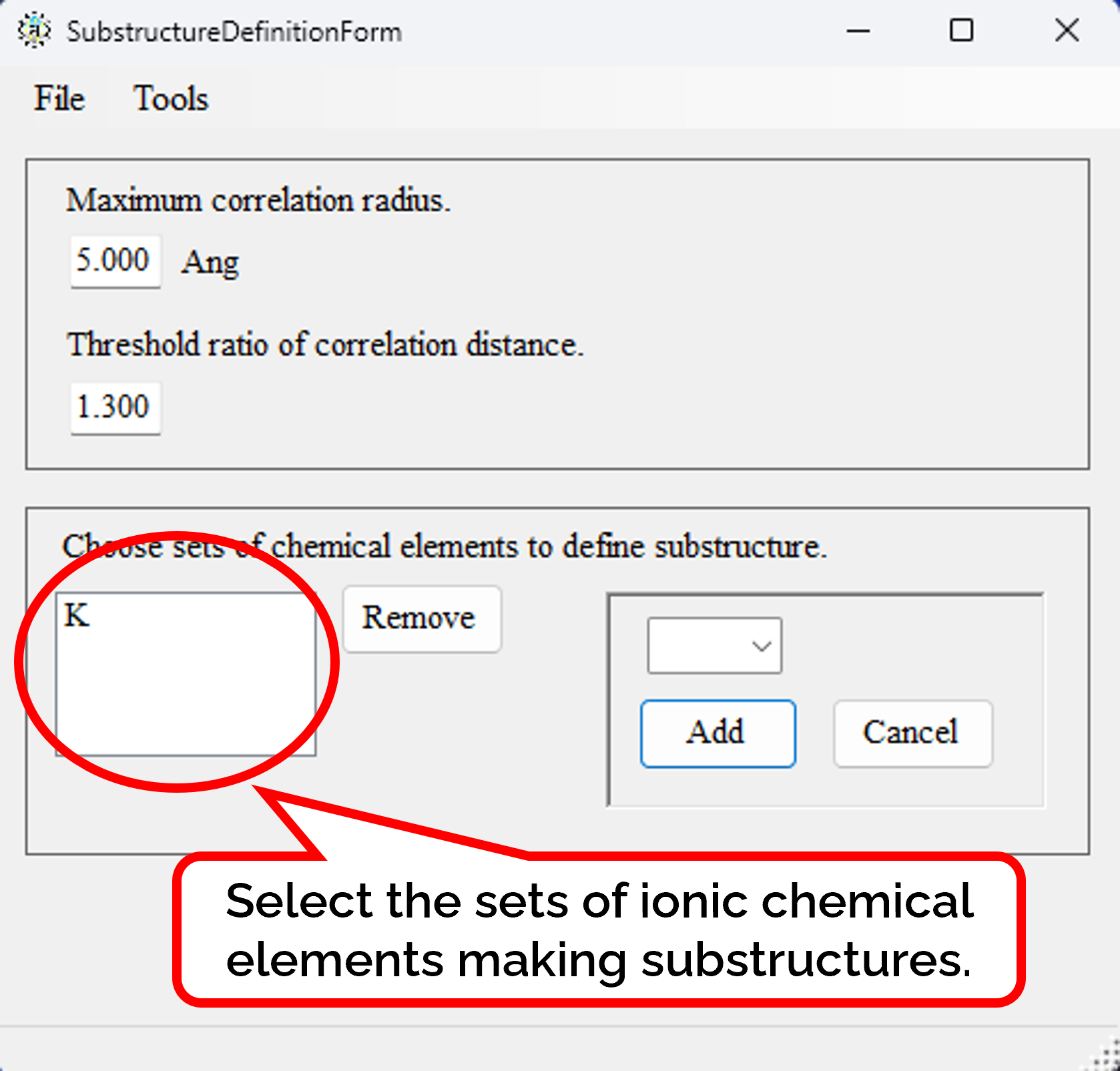
Can crystal structures be classified by their substructures?
Crystal structures can be classified by their substructures. The substructure is made by removing atoms that do not belong to any designated set of ionic chemical element.
Input file generation for execution on Linux
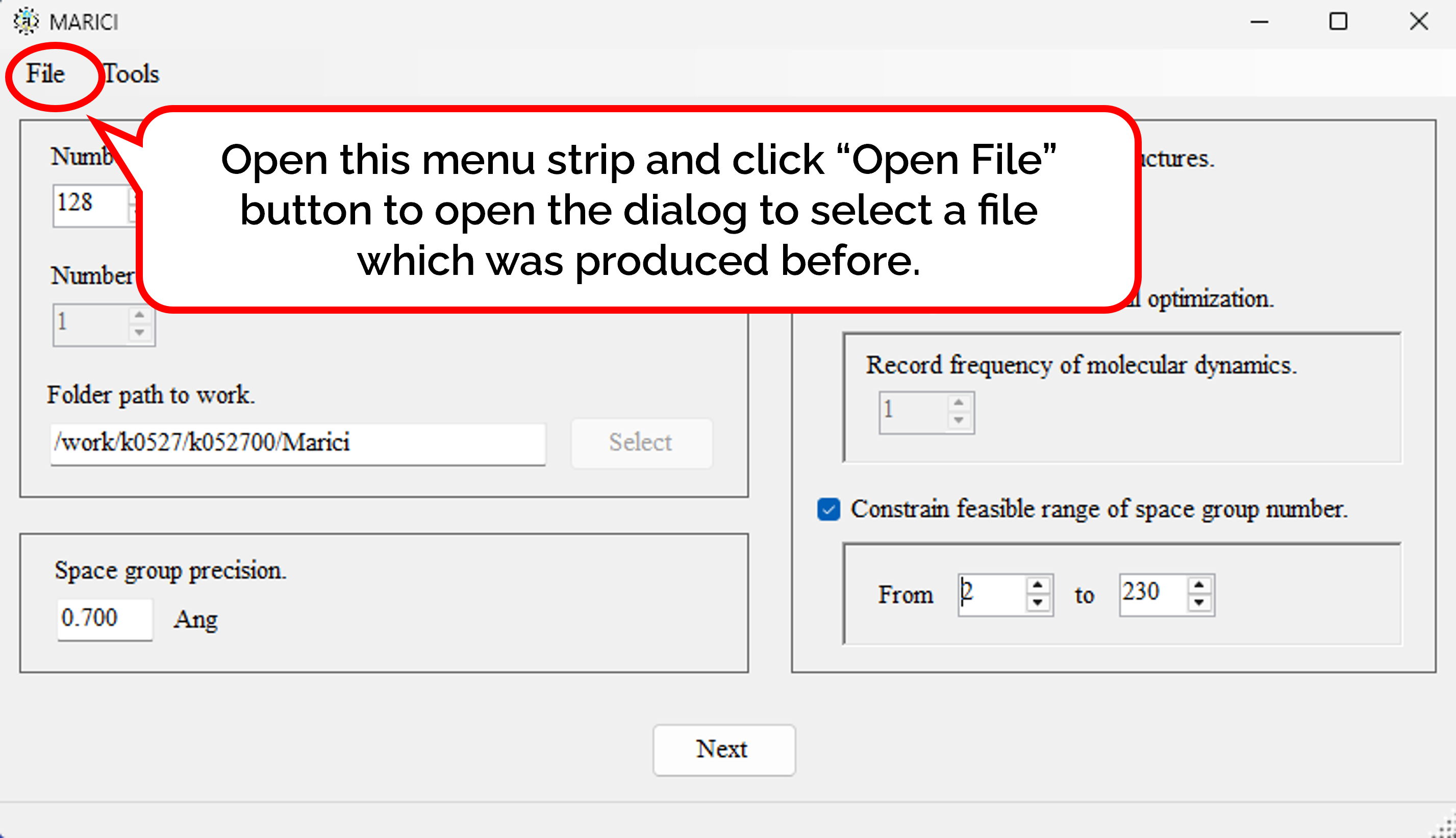
How to copy previous parameters to extract crystal structures?
You can easily copy the previous parameters to predict crystal structures on Linux as follows: First, open the menu strip named "File" and click "Open File". Second, select a file which was produced by MARICI before via a "open file dialog". Accordingly, all the parameters to designate parameters to design crystal structures are copied from the file.
| Design thema | Sample input file |
|---|---|
| Prediction of a quaternary oxide | quaternary_oxide.txt |
| Prediction of four kinds of Perovskite-type structures | perovskite_type.txt |
| Prediction of alpha-Pyrochlore and pyroxene | alpha-pyrochlore_and_pyroxene.txt |
| Predictions of Zintl phases | zintl.txt |
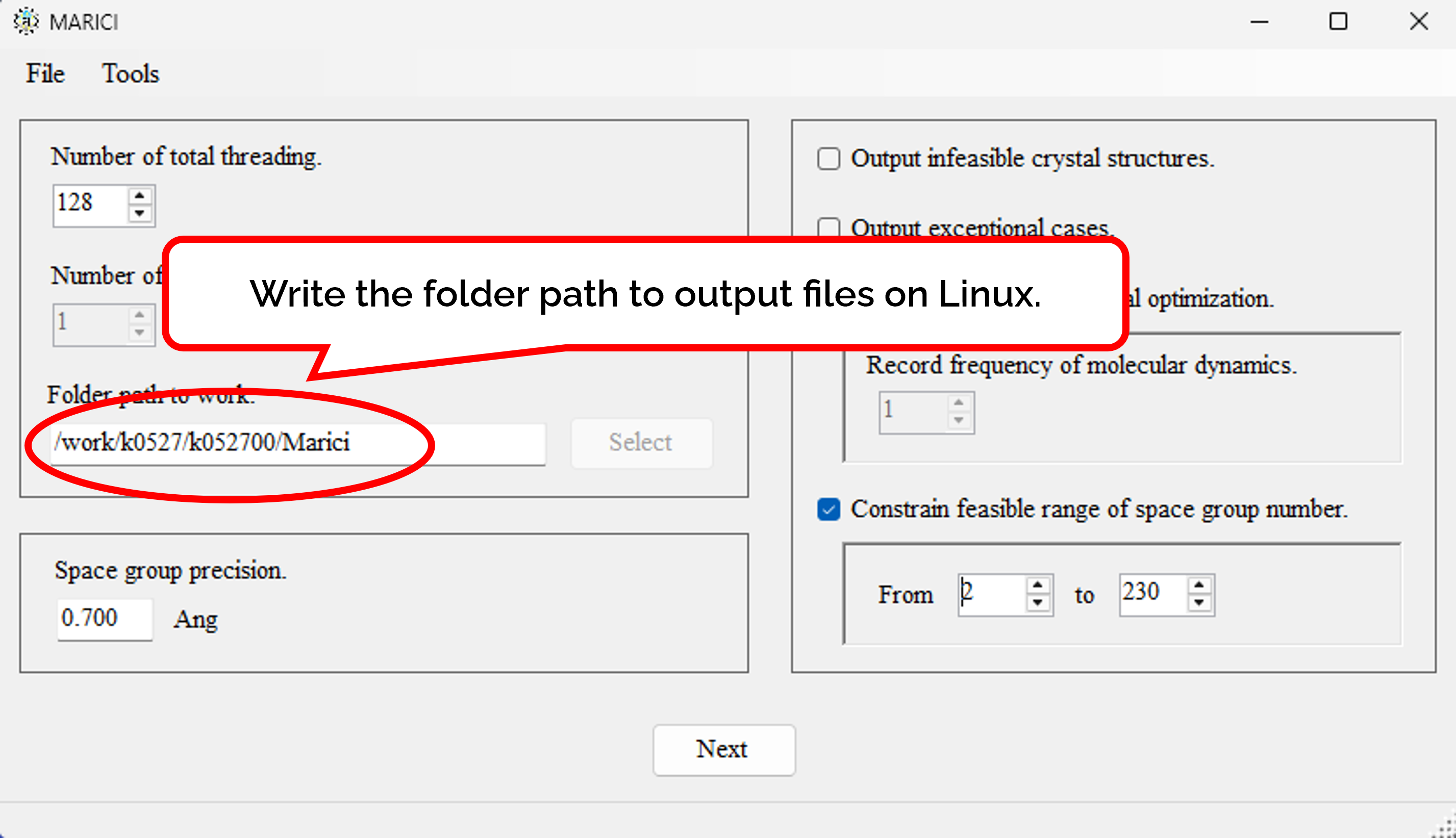
How to designate folder path to output on Linux?
Designate the folder path to output files on Linux in the first form as shown in the figure attached below. The folder path to output files generated by Marici on Windows are designated later.
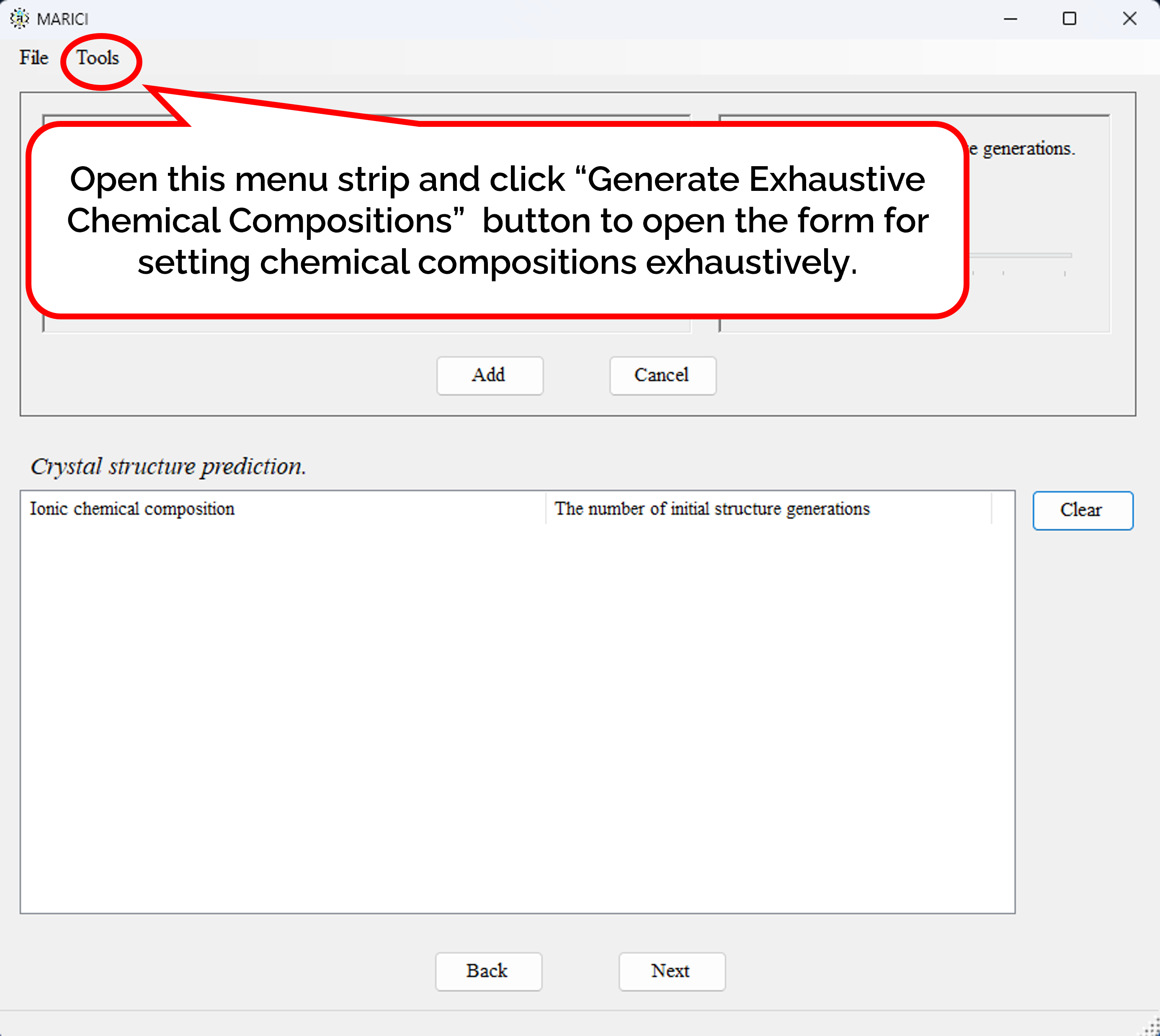
How to set chemical compositions exhaustively?
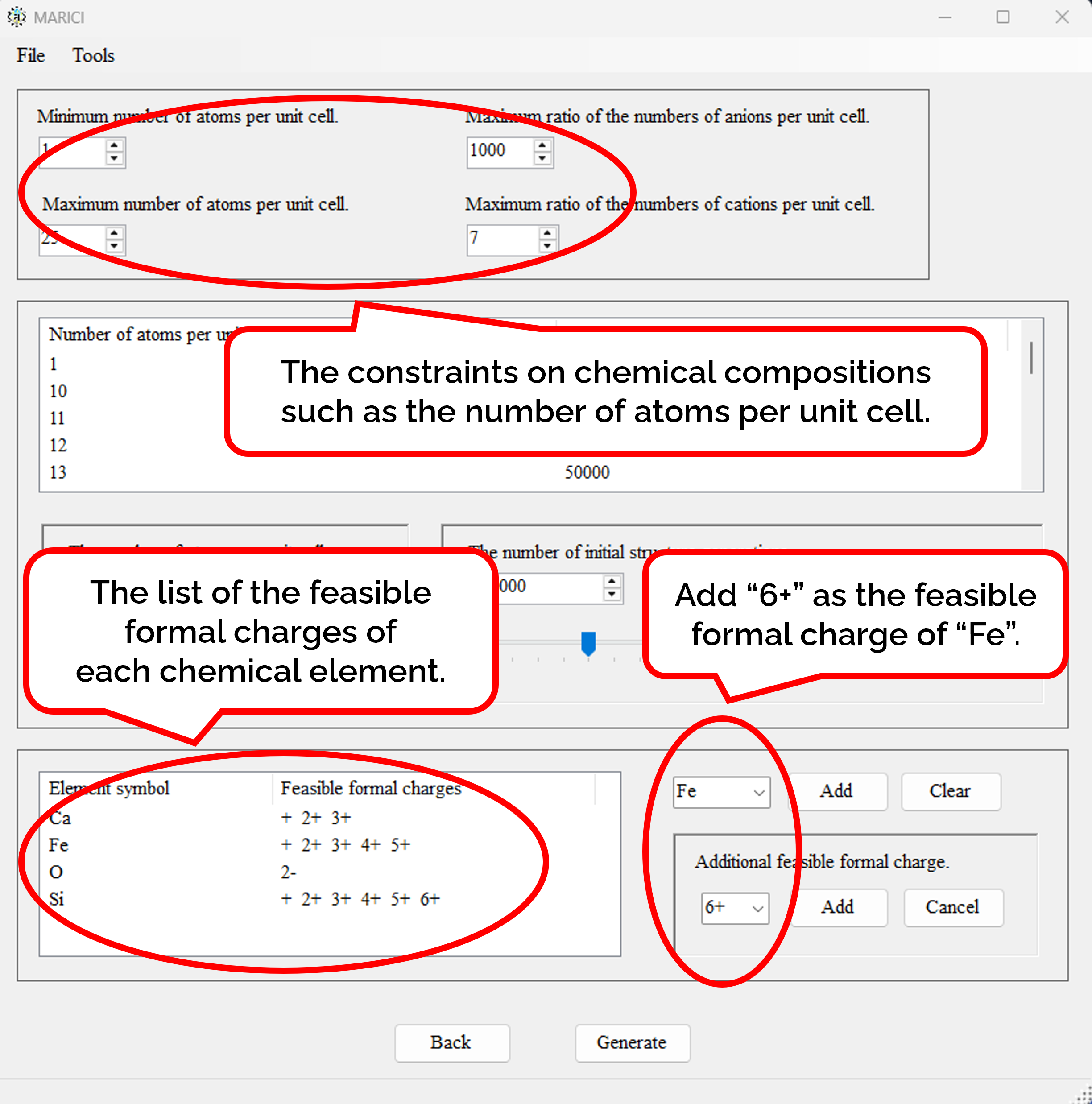
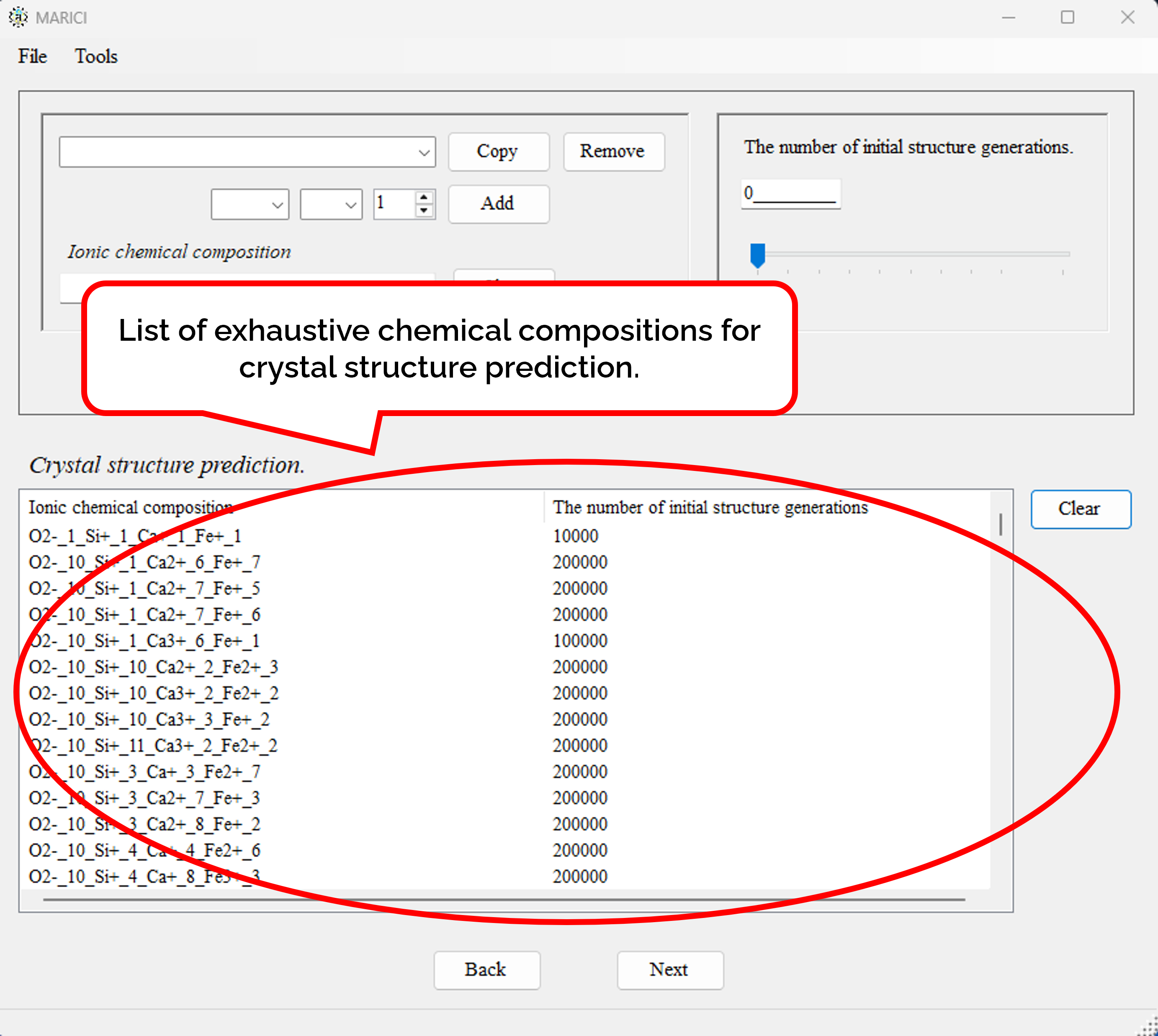
There is the form to generate chemical compositions exhaustively. First, open the file menu and click "Generate Exhaustive Chemical Compositions" button as shown in figure attached below. Second, set the basic parameters to generate chemical compositions such as the feasible range of the number of atoms per unit cell as shown in figure attached below. Third, select the chemical element from the combo box to constitute chemical compositions, and select the feasible formal charges of each chemical element as shown in figure attached below. It is important to note that formal charges are necessary to judge whether an ionic bond are creatable or not between a pair of atoms. Fourth, set the number of randomly generated initial structures depending on the number of atoms per unit cell as shown in figure attached below. Fifth, if you finish setting all the parameters to generate chemical composition, click the next button. When you will go back to the original form to set chemical composition, all the exhaustive chemical compositions are listed with the number of randomly generated initial structures. If the number of chemical compositions is large, it may take more than ten seconds to make the list.

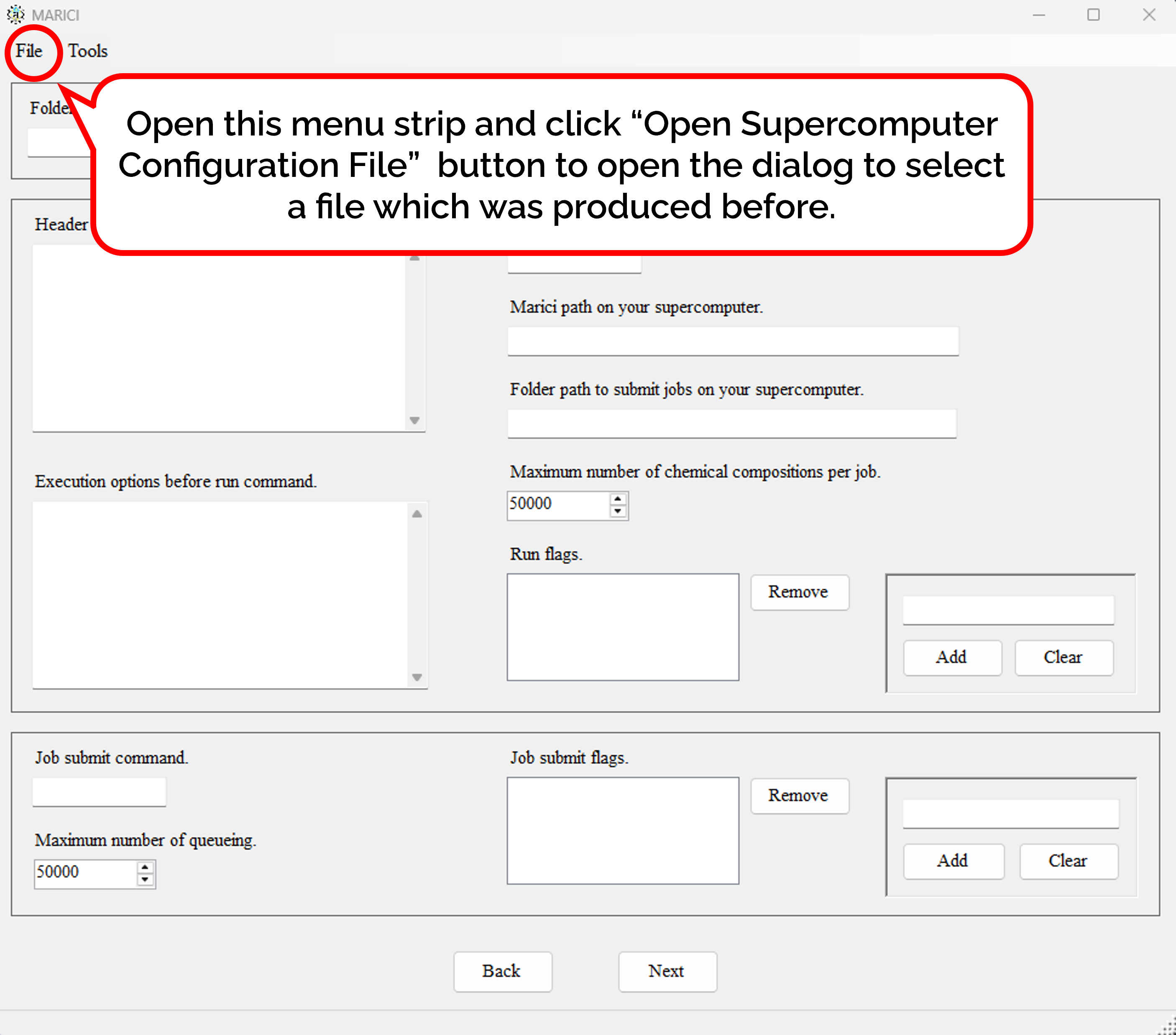
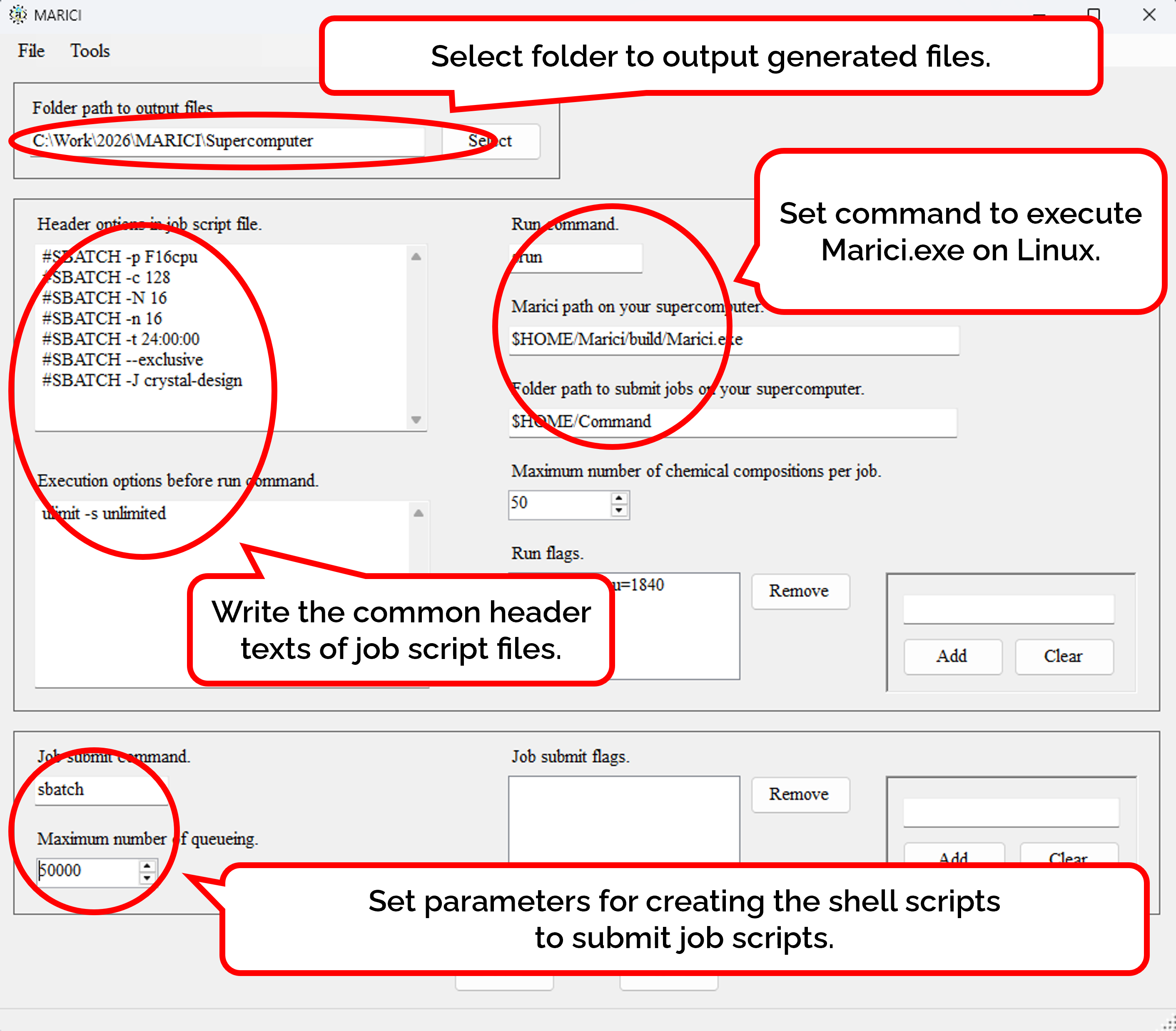
How to generate job scripts for executing MARICI on Linux?
When the number of exhaustive chemical compositions is large, you can separate them into several input files. According to the number of input files, the script files to submit jobs are also generated. As shown in the figure attached below, you can write not only the common header text but also the parameters for making a command to execute MARICI on Linux. Besides, the shell script to submit the job script is also generated. Finally, you can easily copy such parameters from the previous files genrated by MARICI as follows: First, open the menu strip named "File" and click "Open Supercomputer Configuration File". Second, select a file which was produced by MARICI before via a "open file dialog". Accordingly, all the parameters are copied from the file.